Un equipo de investigadores liderado desde la <a href="http://www.uam.es/UAM/Home.htm?language=es" title="Universidad Autónoma de Madrid" alt="Universidad Autónoma de Madrid" target="_blank">Universidad Autónoma de Madrid</a> (UAM) ha identificado un nuevo mecanismo implicado en la patogenia de uno de los trastornos neurológicos hereditarios más comunes, la enfermedad de Charcot-Marie-Tooth (CMT). El hallazgo supone un importante avance hacia el tratamiento y prevención de este tipo de enfermedades.
La enfermedad de Charcot-Marie-Tooth es una enfermedad rara, pero la más frecuente entre las neuropatías hereditarias que afectan a nervios sensitivos y motores. Los pacientes que sufren esta enfermedad presentan una gran debilitación de los músculos inferiores de la pierna, que conlleva deformidades en los pies y pérdida de masa muscular en la pierna.
A día de hoy, se conocen más de 40 genes implicados en la patología. Esto da como resultado una variabilidad en la presentación de los síntomas en los distintos pacientes, y en la afectación de los nervios, presentando variantes desmielinizantes o axonales.
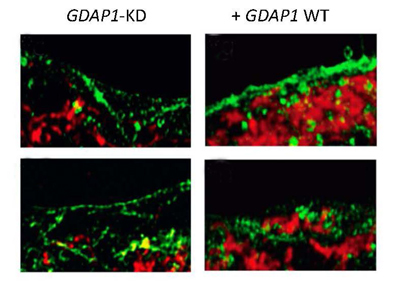
Distribución de la red mitocondrial en condiciones de SOCE activado. Se observa que hay una mayor presencia de mitocondrias (rojo) cerca de la membrana plasmática (verde) en las células de neuroblastoma silvestre (GDAP1 WT) que en las silenciadas (GDAP1-KD).
Ahora, en un estudio publicado en la revista científica Scientific Reports, se ha identificado un posible mecanismo molecular implicado en la enfermedad de CMT causada por mutaciones en el gen GDAP1.
"Este gen codifica la proteína GDAP1 que se localiza en la membrana externa de la mitocondria. Estudios previos indican que juega un papel en la movilización de este orgánulo a lo largo de la célula", explican los autores.
La mitocondria realiza diversas funciones dentro de la célula, como la producción de ATP a través de la respiración celular, el control de la homeostasis de calcio o la producción de especies reactivas de oxígeno. El reciente trabajo presenta un estudio que abarca un amplio número de mutaciones en el gen GDAP1 responsables de distintas formas de la patología.
Los autores identificaron que un mecanismo implicado en el control de la homeostasis de calcio se encuentra afectado en el modelo celular de la enfermedad de CMT. Este mecanismo, denominado Store-operated calcium entry (SOCE), realiza una función esencial en el mantenimiento de los almacenes de calcio celulares del retículo endoplasmático, permitiendo la entrada de calcio desde el exterior celular cuando estos se vacían.
Los resultados obtenidos indican que el mecanismo SOCE se altera como consecuencia de un fallo en la organización espacial de las mitocondrias en células que presentan distintas mutaciones de GDAP1.
"Un hallazgo muy relevante es que las mutaciones afectan de forma diferente al mecanismo SOCE en función de su patrón de herencia, dominante o recesiva, y en función de en qué dominio de la proteína se encuentre la mutación. En particular, las mutaciones recesivas conducen a una disminución del mecanismo SOCE, y a una disminución en el contenido de calcio del retículo endoplasmático", explican los investigadores.
Además, este trabajo pone de manifiesto otras funciones del SOCE hasta ahora desconocidas. La entrada de calcio del exterior, además del mantenimiento de los almacenes de calcio, tiene la función de estimular la respiración mitocondrial, y por tanto promueve la síntesis de ATP celular. Esta función también se encuentra afectada en células que presentan mutaciones recesivas del gen GDAP1.
Los autores del estudio concluyen que el mecanismo patológico que subyace a la enfermedad de CMT causada por mutaciones recesivas del gen GDAP1 podría ser un fallo en el metabolismo energético regulado por calcio.
En el trabajo participan Paloma González-Sánchez, David Pla-Martín y Paula Martínez-Valero, con la supervisión de Araceli del Arco, de la Universidad de Castilla La Mancha (UCLM), y ha sido dirigido por Jorgina Satrústegui de la Universidad Autónoma de Madrid (UAM) en el Centro de Biología Molecular Severo Ochoa (CBMSO), y Francesc Palau del Instituto Pediátrico de Enfermedades Raras (IPER) del Hospital Sant Joan de Déu de Barcelona, ambos pertenecientes al CIBER de Enfermedades Raras.
Referencia bibliográfica:
González-Sánchez P, Pla-Martín D, Martínez-Valero P, Rueda CB, Calpena E, Del Arco A, Palau F, Satrústegui J. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca2+ entry-stimulated respiration. Scientific reports. DOI: 10.1038/srep42993.
