Investigadores de la Universidad Autónoma de Madrid (UAM), junto con colaboradores del Consejo Superior de Investigaciones Científicas (CSIC), han generado y caracterizado un ratón parcialmente humanizado con una mutación frecuente causante de fenilcetonuria
Un equipo de la Universidad Autónoma de Madrid (UAM) ha desarrollado un nuevo modelo de ratón que porta una mutación humana específica causante de fenilcetonuria, una enfermedad rara, metabólica y hereditaria. Dicha mutación afecta el gen Pah, que es crucial para la producción de una enzima hepática que metaboliza el aminoácido fenilalanina.
Dirigido por Lourdes Ruiz Desviat en el Centro de Biología Molecular Severo Ochoa (CBM), y con la colaboración de expertos del CSIC y del Centro de Diagnóstico de Enfermedades Moleculares (CEDEM), el equipo aplicó la técnica de edición genética CRISPR/Cas. Esta técnica permitió reemplazar una sección del gen Pah en el ratón por la secuencia humana afectada por la mutación.
Este nuevo modelo que simula una condición humana (“ratón avatar” de los pacientes con esa mutación), representa una herramienta crucial para la investigación de la fenilcetonuria. Además, el trabajo —publicado en la revista Human Molecular Genetics— facilita la evaluación de nuevas terapias génicas y farmacológicas diseñadas para corregir la mutación humana específica y sus efectos en el ARN mensajero, así como para mejorar la estabilidad y actividad de la proteína mutante resultante.
Ratón avatar de fenilcetonuria
La fenilcetonuria (PKU) es una enfermedad metabólica hereditaria causada por mutaciones en el gen PAH, encargado de codificar una enzima hepática que metaboliza el aminoácido fenilalanina. Detectable mediante la prueba del talón que se realiza a los recién nacidos, la PKU puede provocar retraso en el desarrollo, discapacidad intelectual y problemas de comportamiento si no se trata adecuadamente.
El tratamiento estándar es una dieta baja en fenilalanina, implementada desde los primeros días de vida. Sin embargo, esta dieta frecuentemente enfrenta desafíos, incluyendo el rechazo del paciente y resultados que no alcanzan las expectativas, lo que subraya la necesidad de estrategias terapéuticas mejoradas y más efectivas.
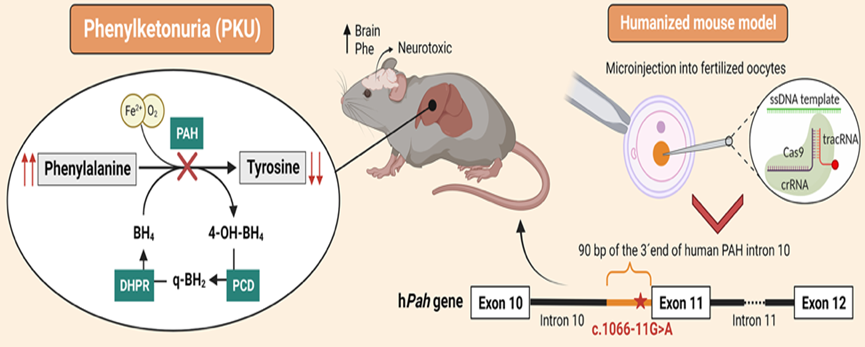
La PKU tiene una prevalencia de 1 en 10,000 nacimientos y se origina por múltiples mutaciones en el gen PAH, que varían en severidad. La segunda mutación más frecuente a nivel mundial es la c.1066-11G>A. Esta mutación causa un defecto en el procesamiento (splicing) del ARN mensajero, dando lugar a una proteína mutante con tres aminoácidos internos extra y, en consecuencia, a una de las formas más graves de la enfermedad.
Esta mutación, especialmente frecuente en países del sur de Europa y en población latinoamericana, es la que ha sido estudiada en el nuevo modelo de ratón PKU. Este modelo de ratón recapitula el defecto molecular de la mutación (alteración del proceso de splicing) y el fenotipo más grave de la enfermedad, con supervivencia y peso corporal reducidos, pelaje hipopigmentado, niveles elevados de fenilalanina, disminución de neurotransmisores y alteraciones locomotoras y de comportamiento (menor actividad y sociabilidad).
A nivel neurológico, los estudios histológicos muestran cambios en la morfología de las células gliales, incremento de marcadores de inflamación y una disminución de la mielinización, detectable también por microscopía electrónica.
A nivel hepático, no hay actividad enzimática PAH, la proteína mutante es prácticamente indetectable, indicando una gran inestabilidad, y se observan niveles reducidos de las chaperonas DNAJC12 y HSP70, y marcadores aumentados de autofagia como LAMP1 y LC3BII, lo que sugiere una posible coagregación de la PAH mutante con las chaperonas y un posterior procesamiento por autofagia.
Estos resultados se reprodujeron en un modelo celular de hepatoma, células HepG2 también editadas mediante CIRSP/Cas con la mutación.
Referencia bibliográfica: Martínez-Pizarro, A.; Picó, S.; López-Márquez, A.; Rodriguez-López, C.; Montalvo, E.; Alvarez, M.; Castro, M.; Ramón-Maiques, S.; Pérez, B.; Lucas, J.J.; Richard, E.; Desviat, L.R. PAH deficient pathology in humanized c.1066-11G>A phenylketonuria mice. Hum Mol Genet. 2024, ddae051. doi.org/10.1093/hmg/ddae051
Fotografía de portada: Imagen de laboratorio donde se ve la diferencia entre los ejemplares, uno más pequeño y de pelaje más claro y otro mayor y más oscuro, arriba a la izquierda. / Lourdes Ruiz Desviat.
